Maladie de découverte récente et encore peu connue, le LPAC syndrome est une prédisposition génétique à la formation de calculs biliaires dans et en dehors du foie. Ce « Low Phospholipid Associated Cholestiasis Syndrome » a été identifié par le Professeur Poupon et son équipe à l’hôpital Saint-Antoine en 2001. Il se manifeste par des douleurs abdominales intermittentes intenses en région hépatique, liées à la présence et/ou migration de calculs biliaires. Son traitement repose sur la prise quotidienne d’acide ursodésoxycholique et nécessite un suivi médical conjoint par le médecin traitant et le gastroentérologue.
Cet article a été rédigé en priorité à l’attention des patients. Pour des informations plutôt à destination du corps médical, nous vous invitons à visiter la page Qu’est-ce que la Lithiase Biliaire (LPAC) ?Read this article in English : Understanding LPAC Syndrome
Pour bien comprendre cette maladie, quelques notions fondamentales sur le fonctionnement du foie et le rôle de la bile
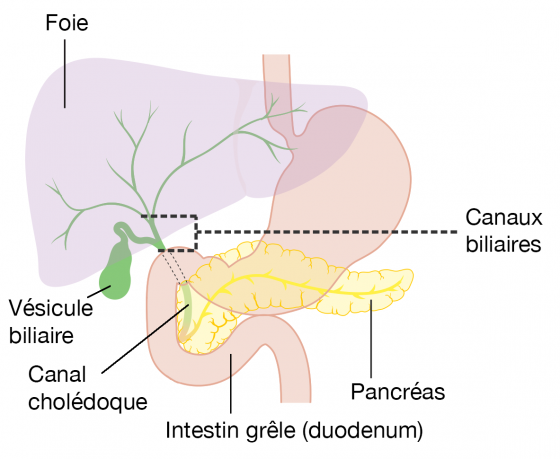
Le foie produit en continu de la bile. Ce liquide verdâtre a 2 rôles principaux :
- digérer les graisses présentes dans notre alimentation, en se mélangeant avec le bol alimentaire dans les premiers centimètres de l’intestin grêle (duodénum),
- se débarrasser des toxines issues du métabolisme du foie, en les évacuant dans le tube digestif, afin qu’elles soient éliminées dans les selles.
La bile est stockée dans la vésicule biliaire pendant les périodes de jeune. L’alimentation stimule la contraction vésiculaire qui se vide alors de son contenu dans l’intestin grêle.
La bile, dans certaines conditions, précipite sous forme de calculs aussi appelés lithiases biliaires. C’est dans la vésicule, siège où la bile à tendance à stagner que ces calculs se forment préférentiellement.
Les lithiases sont très fréquentes : on estime qu’un français sur 5 serait porteur d’au moins une lithiase vésiculaire, et jusqu’à 60% des plus de 80 ans.
Dans 80% des cas, ces calculs sont de types « choléstéroliques », c’est-à-dire composés quasi exclusivement de cholestérol cristallisé. Dès lors, toutes les situations induisant une augmentation du taux de cholestérol sanguin et par voie de conséquence, biliaire, favorisent l’apparition de calculs : l’âge (maximum vers 60-70 ans) ; le sexe féminin, et d’autant plus en période de grossesse (rôle des œstrogènes), le surpoids et les variations importantes du poids ; certains médicaments (notamment pilule contraceptive).
Dans la grande majorité des cas, avoir un ou plusieurs calculs de la vésicule n’a rien d’alarmant, et ce ou ces derniers resteront asymptomatiques tout au long de la vie. Mais 20% des patients avec ces calculs auront des complications biliaires.
Ces complications sont de 4 types :
- la colique hépatique : douleur intense mais de courte durée dans la région du foie (sous les côtes à droite), survenant au moment de la migration d’un ou plusieurs calculs dans les voies biliaires. La douleur est souvent accompagnée de perturbations transitoires du bilan biologique hépatique.
- la cholécystite : inflammation de la vésicule biliaire par « irritation » de la vésicule par la présence de calcul ou par blocage du calcul dans le canal cystique (entre la vésicule biliaire et le cholédoque, qui se jette dans l’intestin grêle). La prise en charge repose alors le plus souvent sur un traitement antibiotique et une chirurgie d’ablation de la vésicule (cholécystectomie) immédiatement ou à distance.
- l’angiocholite : inflammation, voire infection de tout l’arbre biliaire, le plus souvent secondaire à la migration et au blocage d’un calcul à l’abouchement des voies biliaires dans l’intestin. La bile stagne en amont du blocage, causant douleur, jaunisse et fièvre. Le traitement repose sur des antibiotiques et l’ablation du calcul en cause, même si dans la majorité des cas, le calcul finit par s’évacuer de lui-même.
- la pancréatite aiguë : selon le même mécanisme que l’angiocholite, le calcul vient se loger à l’abouchement du canal du pancréas, commun à l’abouchement des voies biliaires.
Tout patient ayant eu un jour une complication liée à un calcul se voit proposer une chirurgie de cholécystectomie (ablation de la vésicule biliaire), afin d’ôter le réservoir des calculs et le mettre à l’abri d’une nouvelle complication.
On peut tout à fait vivre sans vésicule. Cette dernière n’a qu’une fonction de réservoir. Le foie lui, poursuit sa production de bile de façon inchangée.
Le syndrome LPAC (Low-Phospholipid Associated Cholelithiasis)
Le syndrome LPAC est la formation de nombreux calculs biliaires dus à une prédisposition génétique. Il se manifeste par des douleurs soudaines dans la région du foie, et touche principalement les jeunes femmes. Les calculs ne sont pas de même nature que ceux qui se forment fréquemment dans la vésicule, et son ablation chez les malades LPAC est inutile. L’acide ursodésoxycholique est un traitement efficace. Maladie rare et découverte récemment, son diagnostic est souvent difficile.
Qu’est-ce que c’est ?
Le syndrome LPAC est une prédisposition génétique à la formation de calculs biliaires intra- et extra-hépatiques, par anomalie de la composition moléculaire de la bile.
Nous avons vu précédemment que dans le cas de la lithiase biliaire « classique », c’est déjà un problème de composition de bile, trop riche en cholestérol qui induit la formation de calcul.
Dans le LPAC, la bile a une composition normale en cholestérol, mais est pauvre en phospholipides (d’où le nom de low-phospholipid). Ces molécules ont un rôle de solubilisation du cholestérol. En leur absence, il cristallise, formant un calcul.
Une des mutations génétiques identifiées pour expliquer l’absence ou un taux insuffisant de phospholipides dans la bile est celle du gène ABCB4. Ce gène code pour MDR3, un transporteur de phospholipide à la paroi des canaux biliaires. Cette mutation n’est présente que chez 30 à 50% des patients.
La cristallisation du cholestérol dans l’ensemble de l’arbre biliaire induit la formation de nombreux calculs, de localisations multiples (dans la vésicule, mais aussi DANS le foie), et ce dès un âge jeune. Les calculs peuvent ensuite se compliquer, conduisant alors le patient à consulter.
Est-ce une maladie fréquente ?
C’est une maladie de découverte récente (début des années 2000, avec une contribution majeure de l’équipe hépatologie de l’Hopital Saint-Antoine). Cette identification récente explique que nous manquons de données épidémiologiques. On pense actuellement que 1% de l’ensemble des manifestations lithiasiques serait lié à un LPAC. Mais ce chiffre est certainement sous-estimé, il est possible qu’avec l’amélioration de la connaissance de cette maladie, le syndrome LPAC soit diagnostiqué plus fréquemment.
Quel est le profil des patients avec LPAC syndrome ?
Le LPAC syndrome touche surtout des femmes jeunes de 25-35 ans sans surpoids, avec des antécédents familiaux de calculs biliaire.
La présence d’un surpoids n’est pas incompatible avec le diagnostic de LPAC mais le rend moins probable. En effet, le risque de lithiases banales est alors plus important (association surpoids/hypercholesterolémie).
Il existe très fréquemment des antécédents familiaux de lithiases. Malheureusement, également présents chez les porteurs de lithiases banales, ils ne permettent pas d’aider au diagnostic du syndrome LPAC.
Quels sont les symptômes ?
Le LPAC syndrome se manifeste par des douleurs en regard du foie.
Les calculs intra- et extra-hépatiques peuvent rester de nombreuses années asymptomatiques. Quand des symptômes se manifestent, c’est la douleur qui est au premier plan.
D’apparition le plus souvent brutale, pouvant être déclenchée par la prise d’un repas gras et/ou alcoolisé, elle prédomine dans la région du foie (sous les côtes à droites), mais peut irradier dans l’épaule droite. Elle cède habituellement en quelques heures.
La survenue d’autres symptômes : jaunisse, fièvre, ou la persistance de la douleur au-delà de 6h doit faire évoquer une complication liée au calcul, et doit nécessiter une évaluation médicale urgente.
Comment fait-on le diagnostic de LPAC syndrome ?
Le diagnostic repose sur l’âge jeune du début des symptômes douloureux (<40 ans), l’absence d’efficacité de l’ablation de la vésicule biliaire et/ou une échographie hépatique en centre de référence.
Il faut au moins 2 des 3 critères suivants :
- début des symptômes (douleurs de colique hépatique principalement) avant l’âge de 40 ans ;
- récidive des symptômes après cholécystectomie ;
- échographie du foie dans un centre expert, mettant en en évidence des calculs intrahépatiques avec un aspect en « queues de comète ».
La recherche de la mutation du gène ABCB4 peut aider au diagnostic, mais est inconstante (30 à 50% des cas).
Le bilan biologique est le plus souvent normal. En cas de perturbations, aucune anomalie n’est spécifique.
Quelle est son évolution ?
Sans traitement, c’est la récidive des symptômes douloureux qui domine l’évolution de cette maladie.
La présence de calculs dans le foie et dans les voies biliaires expose le patient à toutes les complications lithiasiques présentées plus haut : angiocholite, pancréatite aiguë, cholécystite.
La cholécystectomie, souvent réalisée à tort, ne permet pas d’amélioration clinique. Le risque de développement d’une cirrhose ou d’un cancer hépatique reste à étudier.
Attention au risque de cholestase gravidique, maladie transitoire du 2e-3e trimestre de grossesse, chez les patientes avec LPAC syndrome ou avec mutation du gène ABCB4.
Il ne semble pas y avoir de différence de pronostic selon la présence ou l’absence de la mutation ABCB4.
Comment peut-on la traiter ?
Le traitement du LPAC syndrome repose sur la prise quotidienne d’Acide Ursodésoxycholique (AUDC) à la dose de 10mg/kg. Cette molécule aide l’excrétion de phospholipide dans la bile, restaure l’équilibre dans la composition biliaire et facilite ainsi la solubilisation du cholestérol.
L’AUDC a donc un rôle préventif, en empêchant la synthèse de nouveaux calculs, et curatif, en favorisant la solubilisation des calculs déjà présents.
Ce traitement bien toléré et également très efficace permet d’éviter la cholécystectomie, encore malheureusement proposée en excès. Le traitement est efficace rapidement sur les douleurs (en quelques semaines), plus lentement sur la disparition des calculs intrahépatiques (en quelques mois voire quelques années). Mais l’objectif du traitement reste une amélioration symptomatique.
La durée du traitement reste peu codifiée, mais à envisager à vie.
Aucun régime alimentaire particulier n’est recommandé. Si un régime pauvre en graisse est habituellement prescrit en cas de pancréatite lithiasique, il n’y a pas de données médicales pour le proposer en cas de LPAC. Cependant, des malades rapportent un confort amélioré en adaptant leur alimentation de manière empirique, en fonction de leurs propres réactions.
Comment va se passer mon suivi ?
Le suivi conjoint indispensable par le médecin traitant et le gastro-entérologue est indispensable. Il n’y a à ce jour pas de recommandation pour le rythme et les modalités du suivi de cette maladie.
Néanmoins, le LPAC syndrome justifie d’un suivi médical et notamment gastroentérologique au moins annuel après le diagnostic pour évaluer l’efficacité et la tolérance du traitement.
La surveillance régulière du bilan sanguin, et notamment hépatique peut permettre de déceler la présence de calculs dans la voie biliaire principale en cas de cholestase chronique (élévation GGT et PAL).
Enfin, les jeunes femmes porteuses de LPAC syndrome avec projet de grossesse doivent impérativement avoir un suivi gastroentérologique, car 50% d’entre elles présenteront un tableau de cholestase gravidique, qui expose à un risque d’accouchement prématuré dans 2/3 des cas.
Un autre aspect indispensable est d’effectuer un dépistage familial.
Quel est le risque de transmission à ma famille ?
Cette maladie n’est pas contagieuse. Mais l’origine génétique de cette maladie induit un risque de transmission à la descendance, et ce même en l’absence de mutation ABCB4 identifiée.
A l’heure actuelle, nous ne connaissons pas le mode de transmission (autosomique dominant ou récessif).
Il est important de préciser également qu’il s’agit d’une maladie qu’on dit à «pénétrance incomplète», c’est-à-dire que 2 patients avec la même mutation génétique n’auront pas forcément le même tableau clinique. On peut donc transmettre le gène muté sans que son enfant ne développe de façon certaine la maladie.
Le syndrome LPAC ne justifie pas d’un dépistage prénatal.
Puis-je guérir du LPAC syndrome ?
Le LPAC syndrome est liée à une mutation génétique. Par définition, ces mutations sont présentes à vie. Dès lors, le LPAC vous accompagnera toute la vie.
Le LPAC syndrome est une maladie au pronostic excellent. Le traitement est efficace et bien toléré.
Quelles sont les recherches médicales en cours sur cette maladie ?
- recherche d’autres gènes impliqués dans la physiopathologie de cette maladie.
- mise au point de techniques d’extractions de lithiases intrahépatiques (CPRE)
- études épidémiologiques pour mieux caractériser les patients atteints, et sensibiliser les praticiens à ce diagnostic.

Le livret Syndrome LPAC : comprendre et gérer son quotidien, publié par le laboratoire Mayoly Spindler et l’association albi vous sera peut-être remis par votre médecin, mais vous pouvez le télécharger ici en PDF.